预约国外多囊肾,最快 1 个工作日回馈预约结果
出境医 / 知名医生 / 多囊肾
多囊肾
多囊肾病 (Polycystic Kidney Disease,PKD) 是一种累及肾脏的遗传性疾病,通常有家族遗传性,可呈常染色体显性或隐性遗传,其中以常染色体显性遗传性多囊肾 (Autosomal Dominant Polycystic Kidney Disease,ADPKD) 最常见。流行病学:患病率为 1/1000~1/400病因:多囊肾病的发生与遗传缺陷有关,常染色体显性遗传性多囊肾的突变基因主要有 PKD1 和 PKD2症状:多囊肾病早期可能并没有任何症状,但随着时间的推移将逐渐损害肾脏,出现高血压、腰痛、尿中带血等症状,以及肾功能逐渐减退治疗:目前只能通过控制血压、药物治疗等来延缓多囊肾病的进展,而无法治愈,必要时进行血液透析或肾移植治疗预防:通过向遗传咨询师咨询,可以降低多囊肾病患者遗传给下一代的风险
多囊肾预防
如何预防多囊肾?
- 遗传咨询:如果患有多囊肾并且正在考虑生育,遗传咨询师可以帮助评估将疾病传染给后代的风险
- 保持肾脏健康:尽可能保持健康的肾脏可能有助于预防并发症
- 控制血压,遵医嘱服用降血压药物
- 保持良好的生活习惯,多吃水果、蔬菜、谷物,低盐饮食
- 经常锻炼身体,保持健康的体重
- 戒烟,节制饮酒
就医
什么时候需要及时就医?
出现以下情况,请及早就诊:
- 一级亲属(包括父母、兄弟姐妹或子女)患有多囊肾病
- 出现多囊肾病的一些体征和症状
- 接受医学检查时发现有多囊肾可能
建议就诊科室
- 肾内科
医生如何进行诊断?
医生通过询问患者病史,进行超声检查、分子遗传学诊断、CT 和 MRI 等结果综合判断确诊。
- 超声检查:敏感性高,无放射性,不会造成创伤,经济、简便,是首选的检查方法
- CT 和 MRI:诊断多囊肾时相对更加清晰,但由于检查费用较高,不作首选
- 分子遗传学诊断:检测基因有没有异常,目前多用于囊肿发生前和产前诊断,以及与其他囊肿性疾病鉴别
医生会问患者什么问题?
- 是体检发现的还是已经出现相关症状?
- 症状持续时间?是否有加重?
- 做过哪些检查?
- 家里有没有其他人患有此病?
患者可以咨询医生什么问题?
- 什么是多囊肾病?
- 这种疾病会让我感觉如何?
- 我有什么治疗选择?
- 有没有并发症或后遗症?
- 这种疾病能治愈吗?
- 如果我的肾脏完全停止工作该怎么办?
我的家里人要接受检查吗?
如果我有孩子,他们会得病吗?
我的孩子需要进行基因测试吗?
怎样让下一胎不得这个病?
日常
日常需要注意什么?
- 低盐(高血压或水肿者)、优质低蛋白(肾功能异常者)饮食,避免不健康生活方式
- 保持乐观、积极向上的人生态度
- 定期随访并遵照医嘱治疗
多囊肾介绍
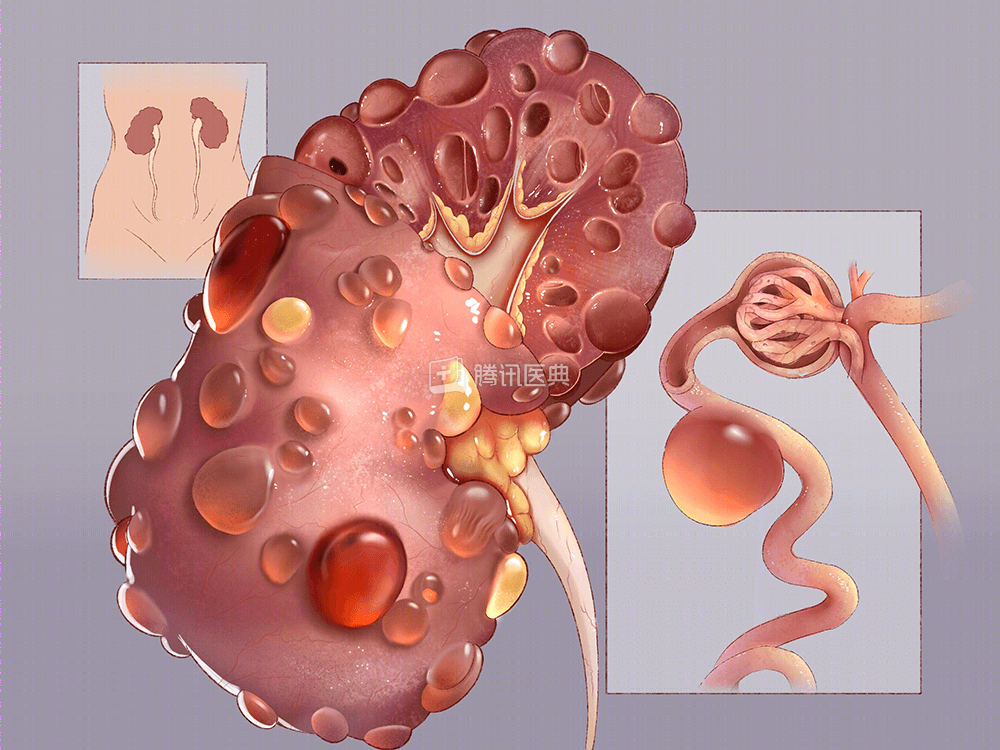
多囊肾病 (Polycystic Kidney Disease,PKD) 是一种累及肾脏的遗传性疾病,通常有家族遗传性,可呈常染色体显性或隐性遗传,其中以常染色体显性遗传性多囊肾 (Autosomal Dominant Polycystic Kidney Disease,ADPKD) 最常见。
- 流行病学:患病率为 1/1000~1/400
- 病因:多囊肾病的发生与遗传缺陷有关,常染色体显性遗传性多囊肾的突变基因主要有 PKD1 和 PKD2
- 症状:多囊肾病早期可能并没有任何症状,但随着时间的推移将逐渐损害肾脏,出现高血压、腰痛、尿中带血等症状,以及肾功能逐渐减退
- 治疗:目前只能通过控制血压、药物治疗等来延缓多囊肾病的进展,而无法治愈,必要时进行血液透析或肾移植治疗
- 预防:通过向遗传咨询师咨询,可以降低多囊肾病患者遗传给下一代的风险
美国多囊肾治疗
如何治疗多囊肾?
常染色体显性遗传性多囊肾为基因突变导致的遗传性肾病,目前尚无特效治疗药物。治疗原则主要为对症处理、预防和治疗并发症、尽最大可能延缓囊肿生长和肾功能恶化。进入终末期肾病时,则进行肾脏透析治疗或者肾移植。
一般治疗
- 限制含咖啡因饮料(如可乐,茶,咖啡等)
- 高血压时限盐
- 病程晚期推荐吃含蛋白质少的食物
- 避免使用对肾脏有毒性的药物
- 当能看的尿中带血时多喝水
- 当囊肿较大时应避免剧烈的体力活动和外力撞击
控制并发症
- 疼痛
- 急性疼痛常为囊肿出血、感染或结石引起,针对病因进行治疗
- 慢性疼痛多因肾脏体积增大所致,当疼痛发作时卧床休息并观察,如果疼痛持续或较重可使用止痛剂。止痛剂不能缓解可考虑手术治疗
- 出血
- 多为囊肿出血所致,会自行好转
- 卧床休息、止痛、适当饮水等保守治疗效果较好
- 对于出血量大,内科治疗无效者,可选择介入治疗或手术治疗
- 高血压
- 感染
- 膀胱炎和肾盂肾炎应选用敏感抗生素治疗,疗程应达 1~2 周
- 囊肿感染时应联合应用水溶性和脂溶性抗生素,必要时局部使用敏感抗生素和引流,囊肿感染一般需要 2 周以上的疗程
肾外症状的处理
- 多囊肝
- 无症状时不需治疗
- 囊肿导致肝脏体积过大时,可引起多种并发症。治疗主要针对减少囊肿和肝体积进行手术治疗
- 脑内动脉瘤
肾脏替代治疗
疾病进展至终末期肾脏病需采用肾脏替代治疗,包括:
- 腹膜透析(通过腹膜作为透析膜进行透析):但增大的肾脏可能影响有效腹膜透析面积,影响腹透效果
- 血液透析
- 肾移植
新型“特异性”药物治疗
近来多项研究显示托伐普坦(精氨酸加压素 V2 受体拮抗剂)可延缓 ADPKD 病人肾脏体积增大和肾功能恶化。可根据病人年龄、肾功能及病情进展情况选用,并注意肝功能损伤、脱水、电解质紊乱等。
导致多囊肾的因素
多囊肾的常见原因是什么?
多囊肾的发生已经明确与遗传缺陷有关,常染色体显性遗传性多囊肾的突变基因主要有 PKD1 和 PKD2。
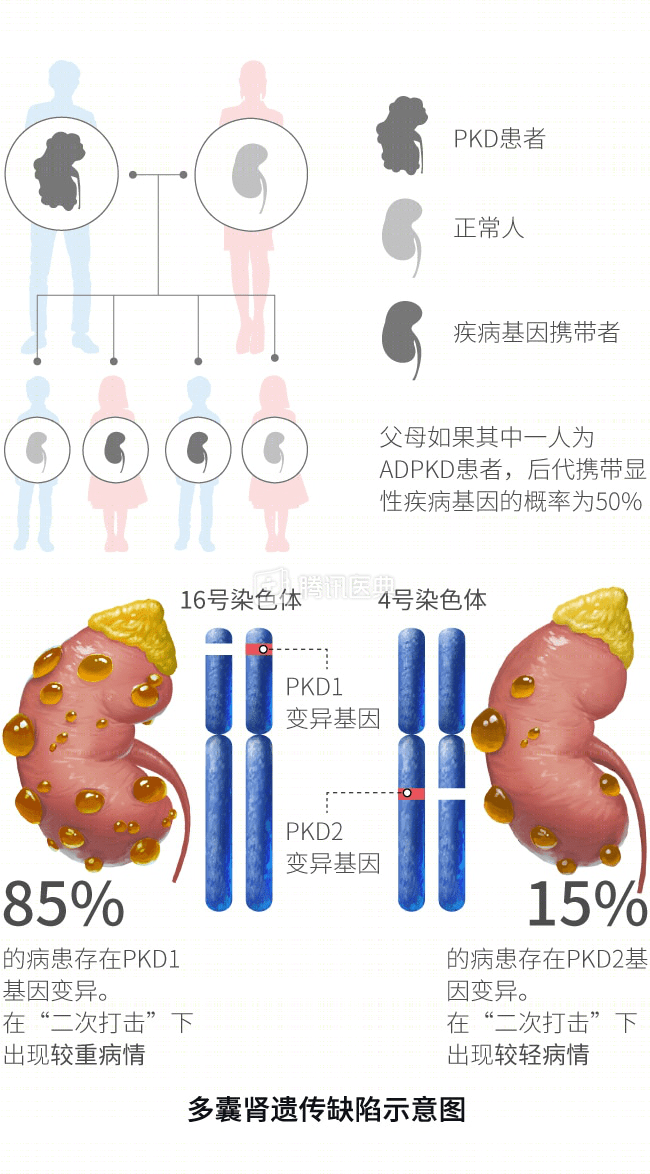
多囊肾的危险因素有什么?
常染色体显性遗传性多囊肾患者出现进展性肾脏病的危险因素包括:
- 遗传因素:PKD2 基因突变患者比 PKD1 基因突变患者进展更缓慢
- 性别因素:男性
- 症状因素:出生体重低,蛋白尿和血尿出现时间早,高血压等
多囊肾症状
常染色体显性遗传性多囊肾病程较长,进展相对缓慢,一般在 30 岁以后出现症状,可损害多个系统,临床表现多样,主要包括肾脏表现及肾外表现。
多囊肾常见的症状有什么?
肾脏表现
- 高血压:是较常见的早期表现,也是促进肾功能恶化的主要危险因素
- 腹部“包块”:体检或患者自己在腹部可触及“包块”,系增大且形态失常的肾脏,也可经 B 超或 CT 发现肾脏结构异常(囊肿形成)
- 背部或肾区疼痛:常为腰背部钝痛、压迫感,有时为腹痛,在肾内囊肿破裂出血或感染时可突发剧烈疼痛
- 血尿:半数患者存在镜下血尿(尿液肉眼观察正常,在医院检查时才会被发现),尿中带血多发生于剧烈运动、创伤后或囊肿血管破裂
- 泌尿道和囊肿感染:主要表现为膀胱炎、肾盂(肾的其中一部分)肾炎、囊肿感染和肾周围脓肿
- 肾功能逐渐下降
肾外表现
美国日本医生

Ricky Clay MD
经验:6-10年

Nathan Formaini DO
经验:6-10年

Sergio Murillo MD
经验:6-10年

Heather Benjamin MD
经验:11-20年

Suzanne Reitz MD
经验:11-20年

Heather Miske DO
经验:11-20年

Bert Hepner DO
经验:11-20年

Steven Paterno MD
经验:11-20年

Aliana Abascal MD
经验:3-5年

Wali Abawi DO
经验:3-5年
临床试验
- Venglustat在中国健康受试者的药代动力学、安全性和耐受性
- GZ/SAR402671 治疗常染色体显性多囊肾病 患者的有效性、安全性研究
- 评估口服 AL01211 在健康志愿者和常染色体显性多囊肾病受试者中的安全性、耐受性、药代动力学和药效学
- ADPKD 中的每日热量限制
- 一项旨在了解 Molidustat 作为治疗日本男性和女性肾性贫血的长期安全性和有效性的研究
- 维生素D吸光度研究 - 纳米液体D3(VIDAS)吸光度的临床试验
- 一项研究以检查过去估计的肾小球滤过率(EGFR)斜率作为慢性肾脏疾病患者快速肾功能下降的风险标志的研究
- 在健康的志愿者和患者人群中成像
- 一项研究,以了解与安慰剂相比,在2型糖尿病和慢性肾脏疾病的患者(重塑试验)(重塑)中,塞米卢皮特如何在肾脏中工作。
- 成人发作糖尿病的新型分类及其与上埃及常见的微血管并发症的关联